


【建模文章解读】采用生理吸收模型考察制剂处方开发中食物效应对低溶低渗弱碱性药物的影响
导 读
本研究通过PBPK建模与模拟,考察和分析化合物X出现食物效应的可能机理。并借助参数敏感性分析功能,发现降低原料药的粒径可降低食物效应,探寻通过制剂手段避免食物效应的可行方案,为制剂开发提供决策指导。通过本案例的学习,类似化合物制剂开发时可考察食物效应的影响。
参考文献
Zhang H, Xia B, Sheng J, et al., Application of physiologically based absorption modeling to formulation development of a lowsolubility, low permeability weak base: mechanistic investigation of food effect. AAPS PharmSciTech. 2014 Apr; 15(2): 400-6. IF: 2.666
案例摘要
生理药代动力学(PBPK)模型已经广泛应用于指导药物的研发工作,本案例建立了化合物X的PBPK模型以系统性地研究该药所观测到的正向食物效应的潜在机理,并探索在制剂开发中避免食物效应的可行策略。
通过模型的搭建与分析,对化合物X出现食物效应的潜在机理进行了合理的解释,并发现降低原料药(API)的粒径可以提高化合物X空腹状态下的口服吸收,进而降低该药食物效应的影响。
软件用途
本案例通过GastroPlus软件搭建化合物X在人空腹和餐后给药后的PBPK模型,通过模型参数的输入和优化,主要PK参数(Cmax, AUC, Tmax)的预测结果均在观测数值的±25%范围内;通过模型的建立和分析,还寻找到该药物出现食物效应的可能机理;借助软件的参数敏感性分析(PSA)功能,发现在临床给药剂量范围内,通过降低API的粒径可以降低其食物效应。
1. 研究背景
化合物 X是一个亲脂性化合物,表现出pH依赖性的低溶解特性以及中等程度的肠道渗透性能,属于生物药剂学分类系统(BCS)的II类或IV类。
化合物 X以速释胶囊制剂上市,其系统暴露会在餐后升高。化合物X 400mg单剂量口服给药,低脂餐给药后,AUC和Cmax分别较空腹给药提高了29%和55%,高脂餐给药后,其AUC和Cmax分别较空腹给药提高了82%和112%。
食物影响药物吸收的机理包括延迟胃排空、改变胃肠道pH以及促进胆汁分泌等。由于进食导致胆酸盐浓度增加而起到增溶作用,亲水性溶解度差的脂溶性化合物通常表现出正向的食物效应(吸收增大),这在其它很多研究案例和文章中有报道。
基于体外生物体相关的溶解度、溶出数据以及所研究的动物体内数据,可以整合到PBPK模型中以定量评估与预测口服药物的吸收和食物效应的情况。该案例采用类似的方法,以理解食物对化合物 X吸收影响的机理,并寻找可行的制剂手段以克服该药物的食物效应。
2. 建模数据与处理
2.1 化合物 X的相关建模参数
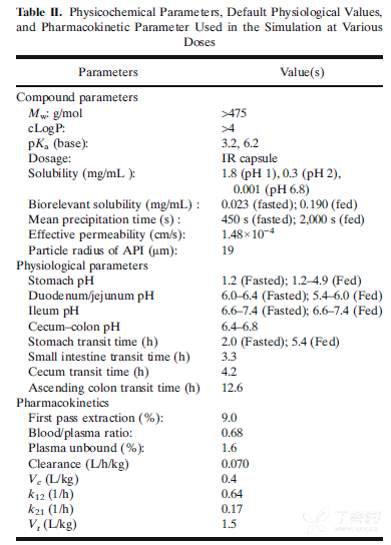
2.2 数据获取及处理
pKa, logP, solubility, Caco-2渗透性等数据均来自体外实验的测定数值;采用文献报道的相关公式,将Caco-2细胞的表观渗透性数值转化为人体有效渗透性(Peff);
为准确考察药物体内的溶解特性,进一步测定了SGF, FaSSIF, FeSSIF等介质下化合物的溶解度并输入到软件模型中;由于没有很可行的方法定量测定沉淀时间,因此化合物 X的体内沉淀时间是基于观测的人体药代曲线进行拟合的数值;
默认人的生理学模型(Opt LogD model SA/V 6.1)选择用于描述药物在人胃肠道的渗透性行为;药物的亲水性及生物体相关溶解度,以及扩散系数、粒径等参数整合到软件的溶出模型(Johnson)中以描述药物在空腹和餐后状态体内的溶出行为;
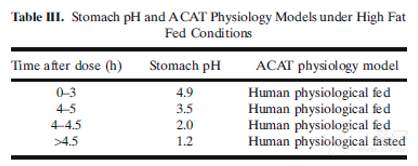
为准确反映食物引起胃中pH值的动态变化,本案例中根据文献报道的结果,将胃pH设置为一条随时间连续变化的曲线,相关的参数设置如下:
人体单剂量空腹口服400mg 化合物 X的物质平衡试验结果表明,大部分的药物(~70%)在粪便中以原型的形式收集,表明该药物在空腹状态下具有较差的口服吸收;
药物的代谢主要发生在肝脏组织,经CYP3A4介导的羟基化和氧化反应;原型药物是血浆循环中的主要成分并产生主要的药理活性;由于缺少合适的静脉给药剂型,未研究人体静脉给药后的体内行为,生物利用度估算为小于30%;
化合物 X人体主要的处置参数(清除率和分布体积)收集于文献验证后的模型数据,其中CL/F的数据根据NCA的计算结果做了微小的调整。
3. 模型结果与分析
3.1 化合物 X 的PK模型的搭建与验证
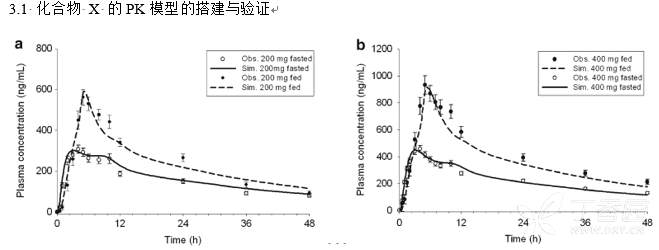
a. 200mg空腹和餐后给药后预测PK曲线与观测点b. 400mg空腹和餐后给药后预测PK曲线与观测点
200mg和400mg 化合物 X空腹和餐后状态下给药后模拟的PK曲线结果如上图所示,模型结果的准确性通过对比预测的PK参数(AUC, Cmax, tmax)与观测数值间的差异进行评估和验证,
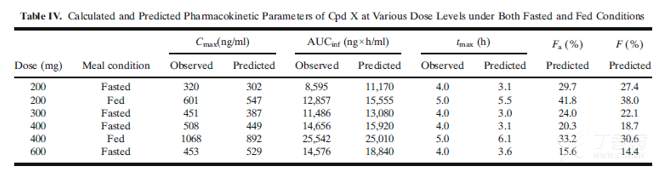
结果表明两种食物状态下,不同剂量的预测结果均在观测均值的±25%范围内,所有剂量的预测与实测结果的PK参数列举在下表:
3.2 化合物 X的体内吸收特征
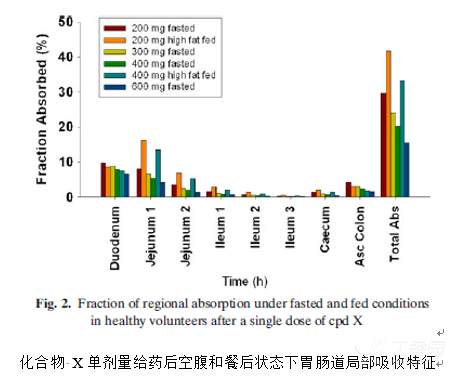
通过PBPK模型预测得到化合物 X在胃肠道不同部位的口服吸收百分数,结果列举在下图。
模型发现该药物的吸收主要发生十二指肠和空肠等上端肠段;回肠中的吸收很少;
此外空腹和进食两种状态下该药物在结肠中都有不少于5%的吸收,这可能是由于该药物在高pH条件下限制了其溶解性能。
化合物 X还展现出了剂量依赖性的吸收,其口服吸收百分数Fa会随着剂量的增加而降低,如空腹状态下给药200 到600mg后,其Fa将从29.7% 降至15.6%;餐后状态下给药200到400mg 后,其Fa将从41.8%降至33.2%。
3.3 化合物 X的体内溶出曲线
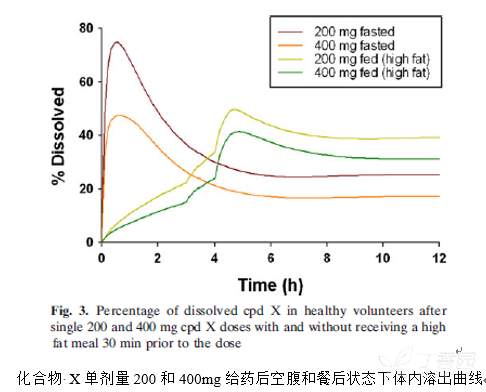
预测得到化合物 X的体内溶出曲线如下图所示。
在空腹状态下,给药后一个小时内药物在胃中快速溶出,然后在到达高pH的肠段部分便快速产生沉淀。
在该状态下剂量从200到400mg时,其溶出的药物量从30%降至20%。
相反,在餐后状态下观察到化合物 X呈现缓慢上升的溶出特征;当饭后3h且胃pH从4.9降至3.5时,此时化合物 X 表现出快速的溶出行为;
所得到的三相溶出曲线,可能提示药物的溶出速率与高脂餐后胃pH的时间依赖性变化有关。此外,餐后状态下药物的沉淀对药物吸收的延迟作用很小。
3.4 化合物 X模型应用-参数敏感性分析
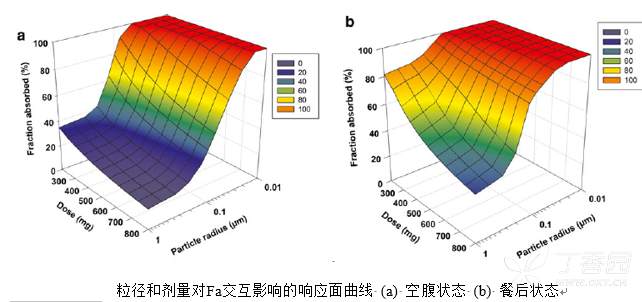
为研究粒径和给药剂量对口服吸收的相互影响,模型开展了空腹和餐后状态下的三维参数敏感性分析(PSA),此外正向食物效应的特征(通过空腹和餐后状态下AUC0-inf的比例进行反映)也基于PSA结果预测的AUC进行了评估。
在空腹状态下,当粒径增加到0.05μm以上时,此时化合物 X的Fa随治疗剂量(200到400mg)的增加急剧降低。此外,在治疗剂量范围内,当粒径不大于0.1μm时该药物的Fa可以提高至60–80%。
餐后状态下,只要粒径小于0.1μm时该药物的Fa均可接近完全吸收(>85%),无论剂量选择在哪个范围(200–800 mg)。当粒径在0.1–1μm范围时,Fa会降低至35–80%,且Fa降低的幅度会随着剂量的增加而加大。
对比空腹和餐后的状态,发现粒径在大于0.1 μm时,该药的Fa仍具有较大的差异。然而,这种差异将伴随药物粒径减小至纳米范围(0.01–0.1μm)时而显著降低。
4. 模型讨论
食物可以通过改变生理条件或者直接产生药物-食物相互作用等方式显著影响药物的吸收和处置行为,通常低溶解度的BCS II或IV类药物会观测到正向的食物效应。
这类脂溶性、弱碱性且呈现pH依赖性溶解特征的化合物,将会在肠道pH条件下表现出低溶解行为并因此产生过饱和而沉淀。
沉淀的药物将进一步聚集成为细小的颗粒,且在高pH值溶解限制的条件下仅有部分能够复溶,会导致空腹状态下药物的不完全吸收。
伴随着进食的发生,药物可以在胃中停留较长时间以延长药物溶出过程,进而提高药物的吸收。此外,进食后分泌的胆酸盐可以显著提高化合物 X在肠道环境中的溶解度,也能进一步提高其口服吸收。
空腹状态下,化合物 X观察到快速且显著的沉淀现象,相反餐后状态则极大地延迟了药物的沉淀;这提示食物的存在可以阻止化合物 X的沉淀发生。
此外,化合物 X 400mg餐后给药后,其tmax从4小时延长到5小时,这说明进食后药物化合物 X可以一直维持着溶解的状态并在肠道下端也有连续的吸收过程。
体内真实的沉淀时间,即过饱和状态的药物在局部浓度超过药物溶解度时从溶解状态产生沉淀颗粒的平均时间,其数值的测定一直是技术难题。
在本案例中,沉淀时间是根据临床获得的一系列血药浓度数据通过PBPK模型拟合得到的。模型最终确定的两组沉淀时间(2000和450s)可以反映餐后和空腹状态下化合物 X体内真实的沉淀速率。
餐后状态能够降低化合物 X的沉淀,可能是因为食物中的脂类成分提高了药物的润湿性,以降低化合物 X固体颗粒与胃肠道液体间的界面表面能,从而提高化合物 X在胃肠道中的溶解特性。
已经报道过胆酸盐可以通过增加药物在胃肠道中的溶解度以提高药物的吸收。该案例中,采用包含胆酸盐和卵磷脂的生物体相关介质测定药物的溶解度,可以评估生理条件下的药物体内溶解特性,这对准确考察药物的吸收和生物利用度提供有用的信息。
模拟过程中,在FaSSIF和FeSSIF介质中测定的化合物 X溶解度一并输入到模型中,以辅助更好地预测其血药浓度-时间曲线。
降低正向食物效应的常用策略是提高药物在空腹状态下的口服吸收,以减小药物在空腹和餐后状态下口服吸收的差异。通常,如果一个药物在空腹状态下有较好的吸收(Fa>70%),临床中观察到正向食物效应的情况不是特别显著,因为进食所起到促进口服吸收的作用有限。
参数敏感性分析结果指示,化合物 X可通过减小粒径的方式消除食物效应的发生:剂量不高于400mg时,保持药物的粒径小于0.1μm以下可以使得化合物 X空腹状态下的Fa大于70%;在更高的给药剂量下,需要将粒径控制在0.05μm以下才能达到相似的口服吸收。因此,化合物 X粒径降低到纳米粒子范围内,对消除化合物 X的食物效应似乎是一种有效的策略。
5. 总结
该工作建立了PBPK模型以阐述化合物 X出现食物效应的机理,该药物低生物利用度的主要原因是空腹状态下药物的显著沉淀而导致的吸收不完全;进食的作用会延长化合物 X 的沉淀时间,并增加药物的体内溶解度。
PBPK模型提示药物的粒径是口服吸收的敏感参数:降低粒径可能会提高药物空腹状态下的口服吸收,同时减少空腹和餐后状态下体内药物暴露的差异。
6. 应用软件与模块
该案例应用的软件是GastroPlus (version 8.0),涉及模块有Base, PBPK。
参考文献
Zhang H, Xia B, Sheng J, et al., Application of physiologically based absorption modeling to formulation development of a lowsolubility, low permeability weak base: mechanistic investigation of food effect. AAPS PharmSciTech. 2014 Apr; 15(2): 400-6. IF: 2.666