


【建模文章解读】受溶酶体捕获的药物(如右美沙芬),其制剂处方的标准制定与体外溶出不相关

导 读
右美沙芬是BCS I类药物,但达峰时间很长。
本案例建立了右美沙芬及其代谢产物的PBPK模型,发现导致药物较长时间达峰是因为药物在肠细胞中受到溶酶体的捕获。
通过对药物在体内ADME过程的正确理解,发现该药物速释制剂的溶出速率及进入肠细胞的吸收速率与该药品的临床疗效无关,制剂开发中应重点观察制剂在体外的崩解行为。
参考文献作者及单位
Michael B. Bolger, Joyce S. Macwan, Muhammad Sarfraz, May Almukainzi, Raimar Lobenberg
美国Simulations Plus公司
美国南加州大学药学院(加利福尼亚州),药理及制药科学
加拿大阿尔伯塔大学,药理及制药科学系
阿拉伯联合酋长国Al Ain University of Science and Technology,药学院
沙特阿拉伯 Princess Nourah bint Abdulrahman University
解读人
陈涛,凡默谷技术部
参考文献
Bolger MB, Macwan JS, Sarfraz M, et al., The Irrelevance of In Vitro Dissolution in Setting Product Specifications for Drugs Like Dextromethorphan That are Subject to Lysosomal Trapping. J Pharm Sci. 2019 Jan; 108(1): 268-278. IF: 3.075
推荐理由
该案例采用机制性口服吸收和药代动力学模型考察了右美沙芬(简称:DEX)及其代谢产物(简称:DXO)体内的ADME过程,探讨和分析了右美沙芬DEX血浆中药物达峰时间较长的机理是由于该药物在细胞内被溶酶体的捕获作用而导致的,而该作用是不受药物剂型因素控制的。
通过对药物机理的正确解释,进一步发现速释制剂的溶出速率以及进入肠细胞的吸收速率与其临床疗效无关。制剂开发中可重点观察制剂在体外的崩解行为,使得溶出的药物超过吸收量即可。
通过案例的学习和理解,可正确搭建药物的PBPK模型,并将药物可能的机理纳入制剂开发的关键影响因素,避免制定的制剂处方质量标准与体内不相关。
软件用途
案例中,利用GastroPlus软件搭建了右美沙芬DEX及其代谢产物DXO的机制性吸收和药代动力学模型,并分析了在涉及与不涉及溶酶体捕获作用时对药物体内PK行为的影响,最终发现溶酶体的捕获作用是导致该药物血药浓度-时间曲线较长达峰时间的因素,并分析了右美沙芬DEX速释制剂的溶出速率以及进入肠细胞的吸收速率与其临床疗效无关;
MembranePlus软件用于模拟体外Caco-2细胞渗透性实验过程,并发现右美沙芬DEX可能受溶酶体捕获的作用;ADMET Predictor软件用于预测药物的理化和生物药剂学性质参数。
案例摘要
该案例的主要目的是建立右美沙芬(DEX)及其代谢产物(DXO)在强代谢(EM)和弱代谢(PM)人群的生理药代动力学(PBPK)模型,并进一步用于评估速释制剂的溶出速率对血药浓度-时间曲线的达峰时间Cmax和血线下面积AUC的敏感性。
体外细胞穿透小室的渗透模拟用于确认溶酶体的捕获作用;GastroPlus软件用于建立右美沙芬DEX的机制性吸收和药代动力学模型。
结果发现只有考虑了溶酶体捕获作用预测结果才能和临床观测数值相吻合:模型发现右美沙芬DEX快速吸收进入肠细胞,然而右美沙芬DXE及其代谢产物却表现出缓慢进入门静脉和血浆,推测很有可能是溶酶体的捕获作用导致的。
案例研究还发现右美沙芬DEX速释制剂的溶出速率以及进入肠细胞的吸收速率与其临床疗效无关。因此,对药物吸收和处置过程所涉及机理的全面分析和理解,对于确认右美沙芬DEX临床制剂的产品技术规范是非常必要的。
1. 研究背景
右美沙芬(DEX)为中枢性镇咳药,主要抑制延脑的咳嗽中枢而发挥作用。该药物在胃肠道中具有高溶高渗的特性,报道可以完全吸收。
然而,该药物达到最大血药血药浓度的时间大概为2.5小时,这比典型的BCS I类药物要缓慢很多。此外,右美沙芬DEX还被描述为可能是被溶酶体捕获的药物。
右美沙芬DEX在体内广泛分布,并在胃肠道和肝脏中快速且广泛地代谢。右美沙芬DEX主要的代谢酶是CYP2D6,该酶具有高度的基因多态性,因此会导致在不同人群体内药物暴露水平的差异。
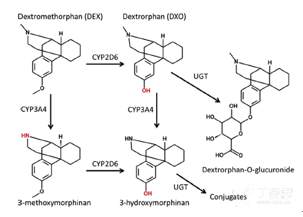
在经历CYP2D6初期O-脱甲基化代谢作用后,其代谢产物去甲右美沙芬(DXO)还将快速共聚成葡糖糖醛酸化产物。此外,DXO还将经历CYP3A4酶作用的N-脱甲基化反应生成3-羟基吗啡烷。DEX也会经历CYP3A4酶作用的N-脱甲基化反应生成次级代谢产物3-甲氧基吗啡烷,并进一步通过CYP2D6作用的O-脱甲基化反应生成3-羟基吗啡烷。
在很多右美沙芬DEX的PK研究中,报道的DXO包括了自由和共聚的DXO。DXO和3-羟基吗啡烷主要以葡萄糖醛酸化共聚物的形式经尿液排泄,其代谢途径如下图所示:
溶酶体是一种单层细胞膜的胞内细胞器,具有低肠道pH (4-5)特点。
溶酶体可分解从外界进入到细胞内的物质;也可消化细胞自身的局部细胞质或细胞器;当细胞衰老时,其溶酶体破裂,释放出水解酶,消化整个细胞而使其死亡。
通过膜H+-三磷酸腺苷酶(ATPase)维持的低pH环境对溶酶体肩负保障许多水解酶最佳活性至关重要。存在与溶酶体腔和细胞质部分的pH梯度,也肩负着某些含氨基结构分子(也称为亲溶酶体药物)的pH分配过程。
有研究报道了亲溶酶体药物的一些典型特征,如logP >2以及介于6.5和11之间的碱性pKa等。
该案例中,采用报道的临床数据和GastroPlus软件搭建了右美沙芬DEX和主要代谢产物DXO+ DXO-O-葡糖糖醛酸总浓度的PBPK模型。
该PBPK模型首先采用一篇研究报道的临床数据进行搭建,然后使用另外一篇文献报道的强代谢(EM)人群的临床数据检测预测的性能。也同时采用了体外Caco-2渗透性模型和机制性口服吸收PBPK模型研究了溶酶体对DEX的捕获作用。最终PBPK模型所提供的详细机理信息以用于评估推荐DEX临床制剂的技术规范。
2. 建模数据与处理
2.1 右美沙芬DEX和DXO的相关建模参数
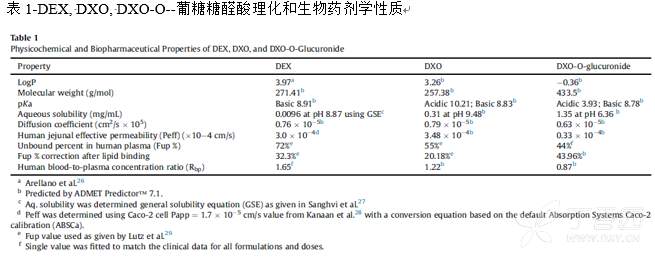
2.2 数据获取及处理
GastroPlus的ACAT和PBPK模型并联合代谢酶与转运体模块以用于搭建右美沙芬DXE的吸收、分布、代谢和消除模型;人体的器官重量、体积、血流速率等数据通过软件的基于年龄的群体估算功能(PEAR)自动产生;药物的理化和生物药剂学性质参数来自于文献报道或使用ADMET Predictor软件预测得到;
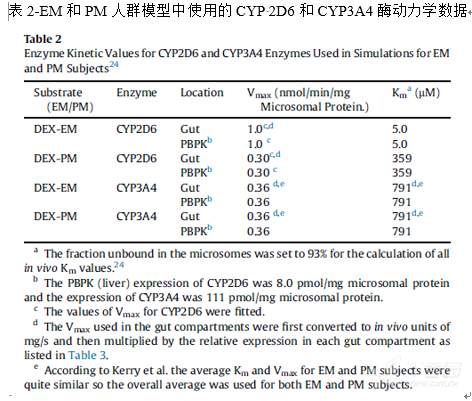
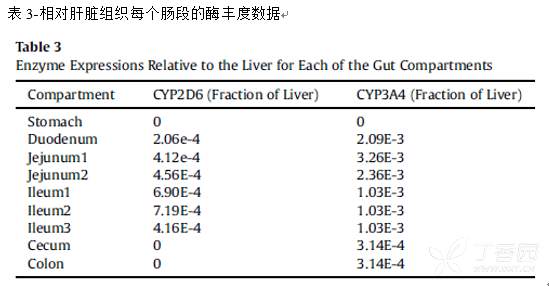
采用GastroPlus软件默认的肠道和肝脏CYP2D6和CYP3A4酶表达数据,其介导的右美沙芬DEX代谢清除是结合酶动力学数据通过米氏方程进行建模计算得到。
CYP3A4的Km数据来自体外,Vmax数据是文献报道参数;CYP2D6的Km数据来自体外,Vmax数据是结合文献报道的EM和PM人群右美沙芬DEX和DXO血浆总浓度-时间曲线进行拟合得到的;拟合的CYP2D6的Vmax数据适合于脱甲基化和葡萄糖醛酸化反应;原型药物的肾脏清除模型是通过Fup*肾小球滤过率来进行指定的;而总的DXO代谢产物(DXO+DXO-O-葡萄糖醛酸)的清除率则是通过估算DXO-O-葡萄糖醛酸化产物的肾脏清除得到,采用肾脏血流量的百分数(0.30)能够很好地吻合代谢产物体内肾脏清除。
在PBPK模型中,药物分布组织中一般有血流灌注型限速和膜渗透型限速。右美沙芬DEX是可以高度渗透至组织中,因此DEX的模型所有组织均设定为灌流限速组织;而对于DXO-O-葡萄糖醛酸具有较差的渗透能力,可能会导致进入机体循环后极低的分布体积,其Vdss是通过Poulin & Theil extracellular方法计算得到的Kp并进一步估算的。
右美沙芬DEX的Kp则是在调整全血-血浆药物浓度比(Rbp) 至1.65后采用默认的机制性Lukacova方法计算得到的,以考虑肝脏、肾脏等组织中溶酶体的捕获作用而导致Vdss的升高。此外,文献报道的游离药物分数(Fup)也参与了模型的计算;
LogP和pKa等参数用于评估药物的处置行为;采用常规的溶解度方程计算了DEX的固有溶解度;体外Caco-2细胞的数值采用软件默认的转化公式得到人体有效渗透性(Peff);药物的理化和生物药剂学性质参数被一起输入到ACAT模型用于模拟药物的体内溶出和吸收特征;
案例中,DEX的分子结构式被输入到MembranePlus软件中,并选择虚拟的24孔板的21-天 Caco-2单层细胞小室模拟其体外渗透特性,以考察右美沙芬DEX可能受溶酶体捕获的作用。
右美沙芬DEX的渗透模型初开始设置浓度为50mM以及75 RPM的转速,分别考察了溶酶体的pH=4.0 (此时,离子型的捕获将会发生)以及pH=6.5两种pH条件,模拟完成后并比较了溶酶体中累积的DEX浓度情况。
肠细胞中溶酶体的捕获作用进一步在GastroPlus软件中通过降低肠细胞游离药物分数(Fuent)至1.3%进行了考察。Fuent的降低能够有效地减少药物在肠细胞内从基底侧膜转运到门静脉的物质量;
采用文献报道的EM或PM健康志愿者的临床数据搭建起了PBPK模型,通过对比预测和观测的速释制剂给药后DEX及代谢产物血药浓度-时间曲线的吻合程度对建立的模型进行了验证。基于PBPK模型所提供的机理解释,可用于研究设定DEX临床制剂的产品技术规范。
3. 模型结果与分析
3.1右美沙芬DEX体外渗透性模型
假设溶酶体的pH分别等于4.0和6.5,并进行了Caco-2细胞小室模型模拟其体外渗透特性,结果列举于下图。
模拟结果提示,供给池和接收池中的药物浓度变化表明DEX是一个典型的高渗透性的药物。当溶酶体pH=4.0时,所模拟的溶酶体中药物浓度比细胞质中的浓度高了4个数量级;而当溶酶体pH=6.5时,溶酶体中的浓度明显下降并仅比细胞质中的浓度高了5倍左右。
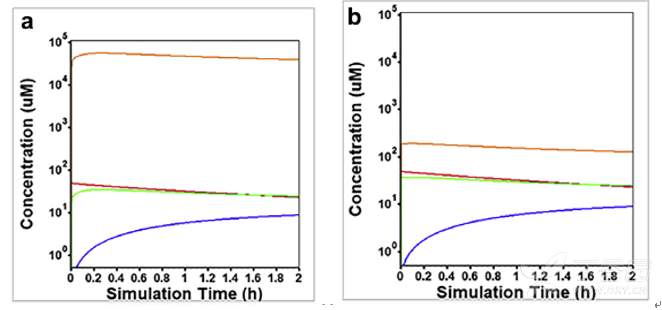
模拟50μM DXE的Caco-2细胞小室渗透实验 (a)溶酶体pH= 4.0 (b)溶酶体pH=6.5;细胞各部分的药物浓度曲线分别为:溶酶体(橙色),细胞质(绿色),供给池(红色),接收池 (蓝色)
右美沙芬DEX受溶酶体捕获作用的图解示意图如下,溶酶体的捕获作用导致了药物在细胞内停留的时间更长,并使得药物从肠细胞进入到门静脉的速率变慢:
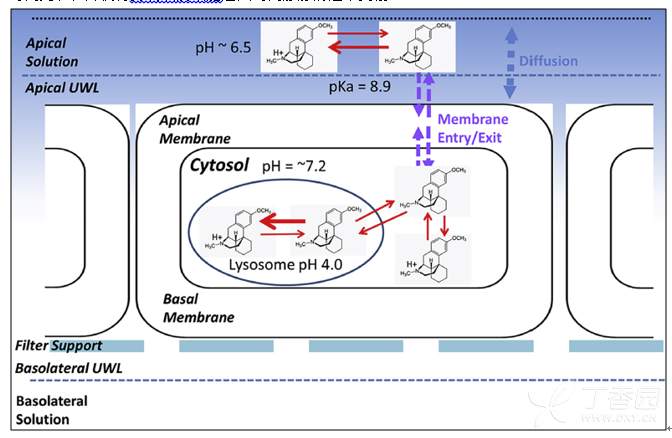
右美沙芬DEX在溶酶体中累积过程的机理解释:右美沙芬DEX具有亲脂和高pKa的特点,药物很容易以非离子状态从肠腔(pH ~ 6.5)跨膜扩散至肠细胞的细胞质(pH ~ 7.2)中,虽然此时药物大部分以离子形态存在。当药物扩散进入溶酶体的酸性环境中(pH 4-5),此时DEX的电离平衡将向DEX离子状态偏移,限制了右美沙芬DEX返回到细胞质中,进而导致右美沙芬DEX在溶酶体中的捕获。
3.2 右美沙芬DEX在PM和EM人群的PBPK模型搭建
右美沙芬DEX在PM和EM人群,以及采用不同Fuent数值考察溶酶体对DEX的捕获作用的PBPK模型采用GastroPlus软件进行了搭建,其预测结果并与文献报道的临床观测数值进行了准确性比较,以确定模型参数和假设机理的准确性,模拟结果图列举如下。结果显示,当Fuent设置为1.3%时能够较准确地接近DEX在EM和PM人群的临床观测数值;而如果把Fuent提高至默认数据100%时,会高估预测的血药浓度-时间曲线。
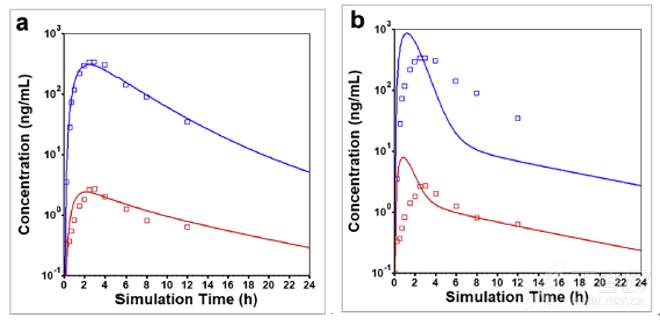
EM健康志愿者口服30mg DEX-HBr速释片剂后观测(点)和模拟(线)的平均血药浓度-时间曲线(a) Fuent=1.3% (b) Fuent=100%;蓝色为DXO总浓度,红色为DEX浓度
所模拟的PK参数与临床观测数值相比,其绝对平均误差(AAFE)基本控制在0.8至1.25范围内(生物等效性范围),DEX及DXO的相关模拟结果汇总于下表:
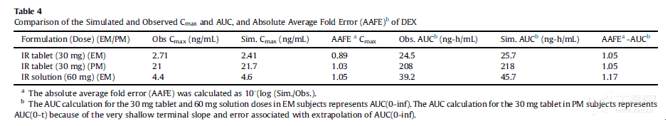
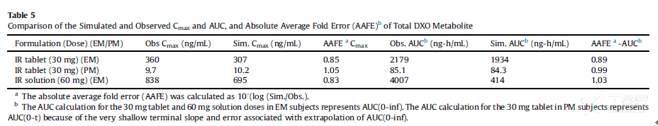
3.3 右美沙芬DEX 在强代谢EM人群的PBPK模型验证
所搭建的DEX 30 mg速释片剂的PBPK模型,进一步预测了文献报道的另一个强代谢EM人群口服给药60mg溶液剂后的PK行为,下图展示了DEX和DXO预测及临床观测的数据。所预测的PK曲线基本与临床结果吻合, 所预测PK参数的AAFE也在0.8至1.25范围内。
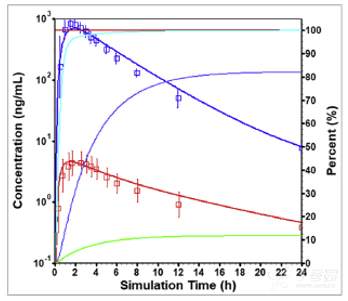
强代谢EM健康受试者口服60mg DEX溶液剂后观测(点)和模拟(线)的平均血药浓度-时间曲线:蓝色为DXO总浓度,红色为DEX浓度。没有观测数据点的红色曲线是体内累积溶出量,蓝绿色曲线是吸收的量,蓝色曲线是进入门静脉的量以及进入系统循环的绿色曲线,所有曲线的右侧Y轴均匀物质的量与给药剂量的百分数进行表示
3.4 右美沙芬DEX模型应用-体外溶出速率对DEX生物等效性的影响
体外溶出一直作为产品技术规范、制剂开发以及确保仿制药生物等效的重要实验,为了分析溶出速率对该类药物不同制剂的生物等效性敏感性,模型进一步考察了粒径的增加对DEX和DXO浓度变化的影响。
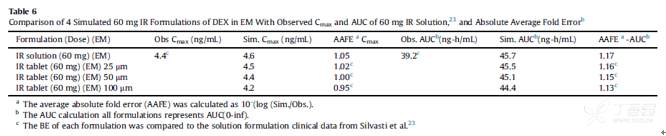
模型从60mg口服溶液剂开始,然后更改为具有不同单分散粒径(25, 50和100μm)的速释片剂,以检测每种制剂与临床观测结果间的生物等效性。所假设的4种制剂中,仅有100μm的制剂(30min溶出83%)体内溶出在30min内低于85%。溶液剂以及25, 50和100μm速释片剂溶出达到85%的时间分别为0, 0.09, 0.25和0.55小时,如下图所示:
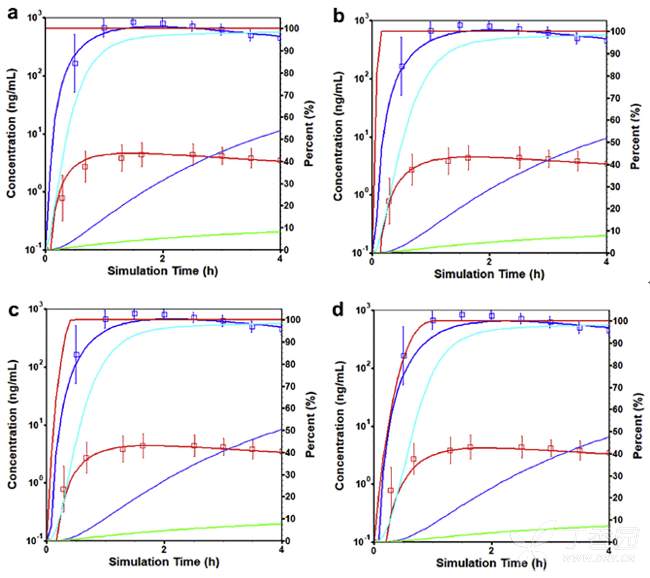
强代谢EM健康受试者口服60mg DEX溶液剂后观测(点)和模拟(线)的平均血药浓度-时间曲线:蓝色带点的为DXO曲线,红色带点的为DEX曲线。
没有观测数据点的红色曲线是体内累积溶出量,蓝绿色曲线是吸收的量,蓝色曲线是进入门静脉的量以及进入系统循环的绿色曲线,所有曲线的右侧Y轴均匀物质的量与给药剂量的百分数进行表示。(a)溶液剂 (b) 25μm速释片剂 (c) 50μm速释片剂 (d) 100μm速释片剂
各个受试制剂所模拟的DEX PK参数的AAFE均在0.8至1.25范围内(PK参数列举在下表),模拟结果表明体外溶出速率对该药的临床疗效及生物等效不敏感。
4. 模型讨论
4.1 右美沙芬DEX在强代谢EM和弱代谢PM人群的肠道首过作用
下图显示了右美沙芬DEX的吸收分数,即药物进入到肠细胞(跨细胞转运)中的量。对于强代谢EM和弱代谢PM人群给药后药物的溶出和进入肠细胞的量基本相似,因为代谢的差异是发生在药物进入肠细胞后。
在肠细胞中,由于药物的理化特性,DEX将经历溶酶体的捕获,使得进入门静脉和体循环的药物相比吸收的药物要明显变慢。但对于强代谢EM和弱代谢PM人群,由于代谢程度的差异使得估算的结果有显著差异。
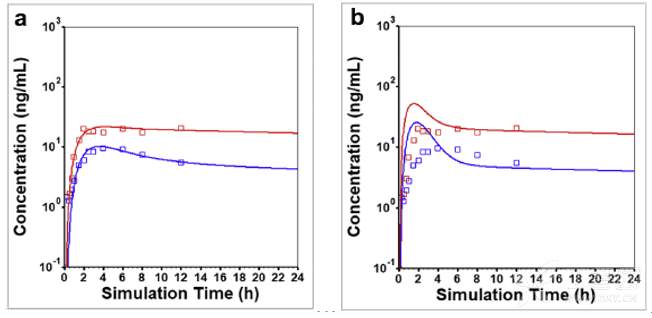
在强代谢EM人群中,DEX有20%会在胃肠道中发生代谢,其余的代谢发生在肝脏中,导致最后仅有14%的DEX进入到机体循环。如果假设Fuent=100%,相比Fuent=1.3%发现在EM人群中会低估肠道的首过效应,进而有更多的DEX进入到门静脉中。
当Fuent设置更小时发现稍大的肠道首过效应可能是因为未达到CYP2D6的代谢饱和,该假设也通过比对设置CYP2D6的Km为1.26mg/mL时肠道的游离浓度进行验证。
当肠道的游离分数设置为100%时,十二指肠、空肠、回肠里平均游离Cmax数据为10.6μM;以及游离分数设置为1.3%,其平均游离Cmax数据为2.3μM。与CYP2D6的Km相比,可以发现将Fuent设置为100%能够更容易产生饱和。
在弱代谢PM人群中,有将近97%吸收的药物可以进一步进入到门静脉中,最后有94%的DEX会进入到系统循环。因此,肠道首过效应对PM人群的作用较少。
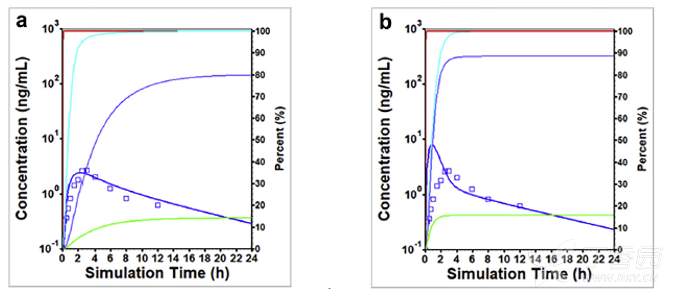
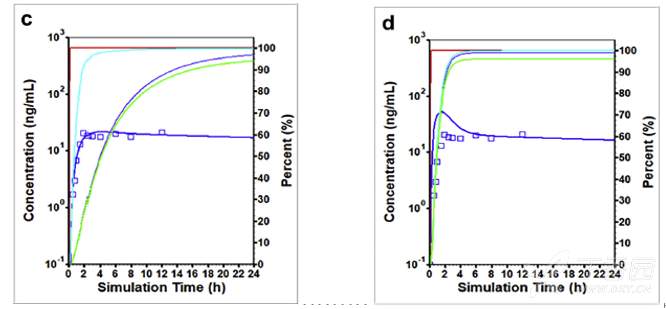
健康志愿者口服30mg DEX速释片剂后观测(点)和模拟(线)的平均血药浓度-时间曲线:(a) 强代谢EM人群且Fuent=1.3% (b) 强代谢EM人群且Fuent=100% (c) 弱代谢PM人群且Fuent=1.3% (d) 弱代谢PM人群且Fuent=100%。
没有观测数据点的红色曲线是体内累积溶出量,蓝绿色曲线是吸收的量,蓝色曲线是进入门静脉的量以及进入系统循环的绿色曲线,所有曲线的右侧Y轴均匀物质的量与给药剂量的百分数进行表示。
4.2 基于PBPK模型设定右美沙芬DEX的产品技术标准
先进的产品技术标准应当反映其临床产品的性能,建立体内外相关性(IVIVC)是构建这种临床相关的方式之一。构建体内外相关的传统方式是将体外药物溶出分数与进入机体循环的百分数建立关系,后者通过Wagner-Nelson和Loo-Riegelma方法将观测的血药浓度-时间曲线进行反卷积计算得到。
与这种方法不同的是,也有文献研究借助右美沙芬DEX的PBPK模型采用血浆数据去拟合药物在胃肠道的溶出曲线。此外,也有采用带入体外溶出数据预测药物体内的PK行为,以间接构建体内外相关性。这些方法都没有考虑药物进入肠细胞的吸收机理,也没有考虑溶酶体的捕获作用导致药物缓慢进入门静脉,以及肠道和肝脏的首过效应。
该工作所搭建的PBPK模型,发现药物能够快速溶出并快速进入到肠细胞,结果提示右美沙芬DEX在体内10min左右能够100%溶出,因此表明该药物的溶出与其临床疗效无关。对于DEX速释片剂,体外崩解实验将更能反映制剂的特性。
在加入溶酶体捕获作用后,所建立的模型能够较好地反映右美沙芬DEX和DXO缓慢进入系统循环的特征。由PBPK模型所提供的机理信息,发现DEX临床制剂的产品技术规范仅需保证药物的溶出超过药物的吸收即可。
5. 总结
该工作所搭建的PBPK模型可以较好地预测右美沙芬DEX和DXO在弱代谢PM和强代谢EM人群的血药浓度-时间曲线,从PBPK模型洞悉的药物机理可以辅助理解药物理化和生物药剂学过程对该药在不同人群体内PK行为的影响。
模型发现溶酶体的捕获作用是导致血浆药物缓慢达峰的主要因素,而此前这种延迟被错误地认为需要设定药物缓慢溶出的制剂产品。然而,临床试验中观测到的药物缓慢达峰现象,主要是因为溶酶体的捕获作用,且该作用是不受剂型因素控制的。
模型结果体现,该药物在胃肠道中的溶出以及进入肠细胞的速率,与其制剂产品的临床疗效是无关的。右美沙芬DEX和代谢产品DXO的血浆暴露水平是不适用于设置溶出性能监测进行评估的。当然,这种对药物吸收和处置过程的整体认知和理解,应当用于确定临床制剂的产品技术规范。
6. 应用软件与模块
该案例应用的软件是GastroPlus (version 9.0),涉及模块有Base, PBPK, Metabolism & Transporter, Optimization, PKPlus, ADMET Predictor;以及ADMET Predictor (version 7.1);和MembranePlus (version 2.0).
参考文献
Bolger MB, Macwan JS, Sarfraz M, et al., The Irrelevance of In Vitro Dissolution in Setting Product Specifications for Drugs Like Dextromethorphan That are Subject to Lysosomal Trapping. J Pharm Sci. 2019 Jan; 108(1): 268-278. IF: 3.075